- General Questions
- Usability Questions
General Questions
What is Clonal Hematopoiesis drivers?
The Clonal Hematopoiesis drivers resource contains genes with signals of positive selection across blood samples of two cohorts of cancer (primary and metastasis). Briefly, the IntOGen pipeline (Martinez-Jimenez et al., 2020) has been applied to somatic mutations identified across the blood samples of each cohort, and genes with significant signals of positive selection in their mutational patterns have been identified.
For specific questions about the IntOGen pipeline, please visit intogen.org.
If you find this resource useful, please cite:
Oriol Pich, Iker Reyes-Salazar, Abel Gonzalez-Perez, Nuria Lopez-Bigas, Discovering the drivers of clonal hematopoiesis, 2020.
How are the blood samples collected?
We downloaded the raw sequences of blood samples drawn from cancer patients as normal paired samples as part of two large tumor sequencing projects: TCGA and HMF. Within TCGA, the blood of primary tumor patients was collected, while within HMF, blood samples from metastatic tumor patients were obtained.
Why is it important to discover all Clonal Hematopoiesis drivers?
Clonal Hematopoiesis is a condition related with the evolution of hematopoietic stem cells either in homeostatic conditions (i.e., associated to aging) or in the face of external selective constraints (e.g., exposure to tobacco carcinogens or chemotherapies). The development of CH is related to several disease conditions, such as arteriosclerosis and heart disease. Genes that are frequent Clonal Hematopoiesis drivers (mostly previously known to be associated to the development of hematopoietic malignancies) have been identified through epidemiological analyses or experimental assays. However, more lowly frequent drivers or genes that drive Clonal Hematopoiesis when faced with external constraints probably still remain to be discovered. Discovering all Clonal Hematopoiesis drivers is important for two reasons. On the one hand, it will allow us to explore multiple mechanisms of Clonal Hematopoiesis in the face of different selective constraints for the hematopoietic stem cells. On the other, it will empower us to better identify Clonal Hematopoiesis across human populations.
How are somatic mutations in blood samples identified?
We developed a “reverse calling” approach that uses the paired tumor sample taken from each patient in the cohort as a reference to call the somatic mutations observed in the blood sample. Several stringent filters are applied to ensure that sequencing artifacts or germline variants are not falsely identified as somatic mutations. This reverse calling approach was applied to the ~12,000 samples integrating the two cohorts.
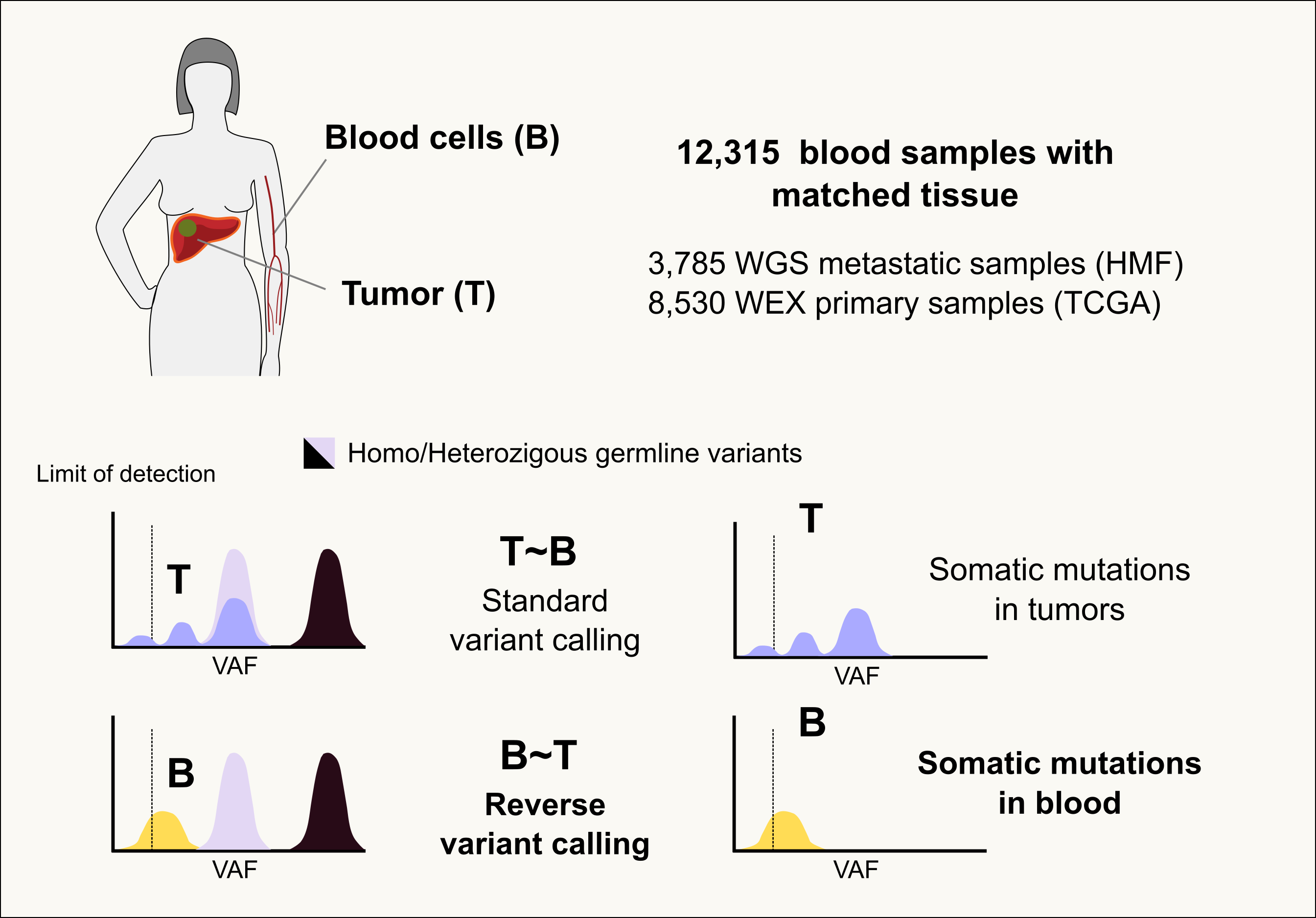
A full description of the reverse calling method is available in Pich et al. Discovering the drivers of clonal hematopoiesis, 2020.
What does it mean that a gene is a known Clonal Hematopoiesis driver?
We compare the list of genes with positive selection signals across the two cohorts with a catalog of a handful of well studied CH drivers (https://doi.org/10.1161/CIRCRESAHA.117.312115). Genes in this list are annotated in the Clonal Hematopoiesis drivers resource as known CH drivers.
What are drivers in IMPACT_blood cohort?
Although the discovery of CH drivers presented in this web was carried out across blood samples collected in the TCGA and HMF projects, we also ran driver discovery methods on somatic mutations detected in targeted sequenced blood samples by the MSK-IMPACT project. Only four of the seven methods in the IntOGen pipeline were run on this cohort, given the hurdle to build a background mutation rate introduced by the targeted sequencing. However, the sample size of the MSK-IMPACT cohort (24,146), being larger than that of TCGA and HMF cohorts lends more statistical power to the discovery of CH driver genes. The genes identified in this analysis are thus labelled drivers in IMPACT_blood cohort to signify the fact that they show signals of positive selection across the blood samples in this cohort.
What is the difference between Clonal Hematopoiesis drivers and IntOGen?
Genes in both resources are identified applying the IntOGen pipeline to a list of somatic mutations observed in genes across patients. Nevertheless, genes in Clonal Hematopoiesis drivers show signals of positive selection in their mutation patterns across the healthy blood of patients, while genes in IntOGen show these signals across somatic mutations in tumors of patients with different cancer types. That is, the Clonal Hematopoiesis drivers resource comprises drivers of Clonal Hematopoiesis and their mutation distribution in blood, while IntOGen comprises cancer driver genes and their mutation distribution across tumors.
Are the results manually curated in any way?
The lists of potential driver genes derived from running the IntOGen pipeline on both cohorts, which are already very enriched for known CH and cancer genes are carefully vetted employing criteria that we have developed in a decade-worth of analysis of cancer cohorts (see the postprocessing section of the IntOGen pipeline documentation). This allows us to rid these lists from likely false positives of the driver discovery methods included in the pipeline. In all, 15 genes are filtered from the union of both cohorts (see Pich et al. Discovering the drivers of clonal hematopoiesis, 2020 for details).
Usability Questions
Which genomic transcript is chosen for each gene?
We use the canonical transcript defined by ENSEMBL as the reference transcript for each gene in our analysis. The current release uses VEP.92 from human GRCh38 genome assembly.
Are you planning to include new datasets?
Yes, we plan to incorporate new cohorts when new cancer datasets become publicly available. We will update the Clonal Hematopoiesis drivers accordingly. Please email us to bbglab@irbbarcelona.org if you have suggestions about datasets that you think could be included.
Can I provide feedback?
Yes, definitely, the resource is still undergoing beta testing. Any feedback is invaluable to us. Please, feel free to drop your comments here: bbglab@irbbarcelona.org.
Why does this site use cookies and what for?
We are using Google Analytics cookies to track usage of our site. CH drivers is a publicly-funded project and these metrics are important to keep support for this project.